OBJECTIVE: To measure muscle strength in patients with spinal muscular atrophy using a handheld dynamometer as an objective tool to evaluate the progression of disease and the outcome of therapeutic trials.
DESIGN: Maximum voluntary isometric contraction was measured in a group of 24 patients aged 5–38 years with types II and III spinal muscular atrophy. Four muscle groups were examined. Data were grouped according to age and sex. Comparison was made between spinal muscular atrophy types; ambulatory vs non-ambulatory, and survival motor neuron (SMN)2 copies. The results were compared with those of a healthy reference population.
RESULTS: Muscle strength was much lower in patients with spinal muscular atrophy than in the healthy population. The walkers group yielded higher values than patients who were non-walkers. Knee extensors were the weakest muscles in both groups, regardless of the ability to walk. The greatest differences were found between ambulatory and non-ambulatory patients. Non-walkers type III patients showed lower values, similar to those for type II patients. Patients with 3 and 4 SMN2 copies showed higher strength with respect to those with 2 SMN2 copies, although not statistically significant.
CONCLUSION: The handheld dynamometer is a valid tool for measuring muscle strength in patients with spinal muscular atrophy. It can be used to measure disease progression and to evaluate changes in therapeutic trials.
Key words: spinal muscular atrophy; muscle strength; weakness; dynamometry; SMN2 copies.
J Rehabil Med 2010; 42: 228–231
Correspondence address: Anna Febrer, Service of Physical Medicine and Rehabilitation, University Hospital Sant Joan de Deu, Passeig Sant Joan de Deu, 2, ES-08950 Esplugues, Barcelona, Spain. E-mail: afebrer@hsjdbcn.org
Submitted June 6, 2009; accepted November 10, 2009
INTRODUCTION
Spinal muscular atrophy (SMA) is a frequent autosomal recessive disorder that affects spinal cord motor neurones, resulting in progressive muscle degeneration and weakness. Severity of SMA varies; it is classified into 3 types according to age at onset of symptoms and motor milestones. Clinical manifestations of type I disease appear in the first 6 months of life; patients are unable to assume the sitting position. Onset of type II SMA occurs after 6 months; the sitting position is possible without assistance. Type III occurs after 18 months of age; patients are able to walk. As the severity of type II varies considerably, ranging from patients who are able only to sit to those who can stand up with orthoses, a number of sub-types have been established (1, 2). Approximately 90% of patients with any type of SMA lack both copies of the SMN1 gene (3). The SMN2 gene is the highly homologous SMN1 copy that is present in all patients, although its presence does not prevent the disease. There is evidence that the number of SMN2 copies acts as a phenotypic modifier. Acute type I patients have 1 or 2 copies and chronic type II and III patients usually have 3 or 4 copies. However, this correlation is not absolute, given that 2 or 3 SMN2 copies have been described in all forms of SMA (4).
Weakness is usually symmetrical, and the proximal muscles at the scapular and the pelvic girdle are the most affected. Moreover, muscles of the lower extremities are weaker than those of upper extremities and the extensors are weaker than the flexors. Thus, joint deformities are often produced in the flexion position (5).
In some type II and most type III patients, the loss of strength is gradual and patients may be stable for long periods. There are also individual variations in type III patients in terms of ability to walk (5). In certain patients with SMA with greatly reduced strength, a functional scale, such as the Hammersmith Functional Scale, is necessary (6–8).
The objective evaluation of muscle strength measurements is complex, particularly in young children. Measurements have traditionally been carried out using the manual muscle test developed by the Medical Research Council (MRC) (9). However, this test is subjective, as results vary among examiners and, moreover, it does not detect small changes in strength. In view of these shortcomings studies have been performed using handheld dynamometers (HD), thus allowing quantification of small changes in muscle strength (10–13).
Because of the advances in the knowledge and therapeutic alternatives in SMA, early clinical trials of candidate therapeutics are now ongoing in patients with SMA (14). Clinical trials in this disease present a unique set of challenges, including the development of meaningful outcome measures, such as validation of motor functional scales and reliable measurements of muscle strength.
We measured muscle strength with HD in a group of patients with types II and III SMA confirmed by genetic analysis of the SMN1 gene. The aim was to validate this tool and to establish associations with age, gender, SMA type, walking ability and SMN2 copy number.
METHODS
Participants
The study participants were 24 patients over 5 years of age who had SMA type II or III defined by the established criteria (1). All the patients had genetically confirmed diagnoses. Three measurements were performed: an initial test, followed by repetitions at 6 and at 12 months. The patients were followed up for 1 year. The study was approved by the ethics committees of the 2 hospitals (HSJD and HSP) collaborating in this work. Informed consent was obtained from the parents of each child, and from adult patients.
SMN1 and SMN2 genotypes
Molecular pathology of the SMN1 gene in our group of patients was determined as described previously (3). To establish the copy number of SMN2, we employed a real-time polymerase chain reaction (PCR) assay based on a specific amplification of exon 7 of the SMN2 gene, as reported elsewhere (4).
Instrument and technique
We used the HD to measure quantitatively, in Newtons, the maximum voluntary isometric contraction in 4 muscle groups. The device was a model CT3001 CITEC (0–500 N) (CIT Techniques BV, Groningen, The Netherlands). The muscle groups studied were knee flexors and extensors, elbow flexors, and the manual grasping muscles. Hip flexors and extensors were not studied because of contractures that are usually present in non-ambulatory patients. The positioning of the patient during examination was established in line with the methodology described by various authors (10, 15). Knee flexors and extensors were evaluated with the patient seated on a gurney with the hip and knees flexed at 90º. The HD was placed distally anterior to the tibia to measure the extensors, and distally posterior to the tibia to measure the flexors. For upper extremities, the patient was placed in a supine position with the shoulder drawn in and the arm at 90º in a neutral position. The HD was placed anterior to the wrist to measure elbow flexors. Manual grasping was determined with the patient seated at a table and with the forearm resting on the table in a semi-prone position. The “make test” technique was applied (16) and 3 values were taken; the highest score was recorded.
Parameter definitions
Muscle strength was studied in association with the following parameters: gender, age, SMA type (II and III), SMN2 copy number and ability to walk at the time of the study. To evaluate age, 2 groups were established: group A with children 8–17 years old and group B with adults 18–38 years of age.
Statistical analysis
Statistical analysis was carried out with the SPSS 14.0 package. Groups were compared according to gender, age group, ambulatory/non-ambulatory and SMN2 copies. Differences were established using the Student’s t-test for independent data. To determine the effect of evolution over time (initial test and final test at 12 months) on muscle force Student’s t-test for matched/paired data was used. Confidence intervals (95%) were based on the t-tests and, in all cases, p-values of less than 0.05 were considered significant.
RESULTS
Of the total 24 patients studied, 15 (62%) had type II and 9 (37%) developed type III disease. The patients’ mean age was 16 years, and age range 5–38 years. There were 15 males and 9 females. Only 3 patients were able to walk during the study: all were males aged 16–17 years. At the time of the study they walked without difficulty, but running was problematic. None of them presented scoliosis, respiratory problems or joint deformities. The 21 non-ambulatory patients presented varying degrees of joint deformity. Fourteen patients had scoliosis, while 6 had been operated on. Six of the type II patients required non-invasive assisted nocturnal ventilation. The group of 9 patients with type III SMA presented normal psychomotor development until loss of ambulatory ability, which occurred between the ages of 7 and 15 years. Eighteen patients had 3 SMN2 copies, 1 had 4 SMN2 and the remaining 5 had 2 copies.
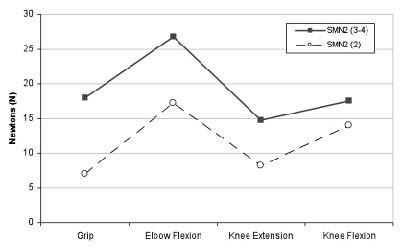
The results of the muscle strength tests showed no statistically significant differences between sexes or age groups (children and adults) (Tables I and II). Knee extensors were weaker than flexors. Muscle groups in the lower extremities showed less force than the elbow flexors, even in ambulatory patients (Table III). Comparison of ambulatory patients with those who had either lost the ability to walk or had never developed it showed statistically significant differences for all muscle groups studied, except for knee flexors. Differences were also significant among the same groups when type III patients who had lost the ability to walk were compared with the ambulatory group (Table IV). Patients with 3 and 4 copies showed higher strength values than those with 2 SMN2 copies, although this did not reach statistical significance (Table V and Fig. 1). No significant differences were found between the values measured at the start and end of the study (Table VI).
| Table I. Force, in Newtons (mean, p-value and confidence interval (CI)), measured with a handheld dynamometer, in 24 patients with type II and III spinal muscular atrophy. Comparison of sexes |
| | Men (n = 15) Mean n | Women (n = 9) Mean n | p | 95% CI |
| Grasping | 19.0 | 9.6 | 0.415 | –32.75–14.08 |
| Elbow flexors | 23.9 | 23.5 | 0.968 | –19.85–19.10 |
| Knee flexors | 16.2 | 12.4 | 0.429 | –13.43–5.92 |
| Knee extensors | 14.1 | 10.2 | 0.400 | –13.42–5.58 |
| Table II. Force, in Newtons (mean, p-value and confidence interval (CI)), measured with a handheld dynamometer, in 24 patients with type II and III spinal muscular atrophy. Comparison of age groups |
| | Group A: Children (n = 16) Mean n | Group B: Adults (n = 8) Mean n | p | 95% CI |
| Grasping | 11.7 | 23.0 | 0.562 | –54.29–31.79 |
| Elbow flexors | 24.5 | 22.3 | 0.834 | –19.50–3.83 |
| Knee flexors | 12.8 | 18.6 | 0.384 | –19.89–8.39 |
| Knee extensors | 11.3 | 9.3 | 0.299 | –4.74–14.66 |
| Table III. Force, in Newtons (mean, p-value and confidence interval (CI)), measured with a handheld dynamometer, in 24 patients with type II and III spinal muscular atrophy. Comparison of ambulatory and non-ambulatory patients |
| | Ambulatory (n = 3) Mean n | Non-
ambulatory (n = 21) Mean n | p | 95% CI |
| Grasping | 83.6 | 5.7 | 0.001 | –101.39 to –54.41 |
| Elbow flexors | 71.6 | 16.9 | 0.013 | –83.83 to –25.60 |
| Knee flexors | 39.3 | 11.2 | 0.069 | –61.26 to 5.16 |
| Knee extensors | 36.6 | 9.0 | 0.001 | –37.22 to –18.11 |
| Table IV. Force, in Newtons (mean, p-value and confidence interval (CI)), measured with a handheld dynamometer in 9 patients with type III spinal muscular atrophy. Comparison of ambulatory and non-ambulatory patients |
| | Ambulatory (n = 3) Mean n | Non-ambulatory (n = 6) Mean n | p | 95% CI |
| Grasping | 83.6 | 11.3 | 0.014 | –124.83 to –19.84 |
| Elbow flexors | 71.6 | 20.6 | 0.007 | –76.51 to –25.48 |
| Knee flexors | 39.3 | 17.1 | 0.101 | –53.80 to 22.57 |
| Knee extensors | 36.6 | 10.6 | 0.004 | –40.82 to 11.18 |
| Table V. Muscle strength, in Newtons (mean, p-value and confidence interval (CI)), and association with SMN2 copies |
| | Two SMN2 copies (n = 5) | Three or more SMN2 copies (n = 19) | p | 95% CI |
| Grasping | 7.0 | 18.0 | 0.250 | –30.34–8.34 |
| Elbow flexors | 17.2 | 26.7 | 0.209 | –25.29–6.11 |
| Knee flexors | 14.0 | 17.5 | 0.520 | –15.12–7.96 |
| Knee extensors | 8.2 | 15.6 | 0.086 | –14.18–1.02 |
| Table VI. Force, in Newtons, (mean, p-value and confidence interval (CI)), measured with a handheld dynamometer, in 24 patients with type II and III spinal muscular atrophy. Comparison of values at initial test and final test, 12 months later |
| | Initial test Mean n | Final test Mean n | p | 95% CI |
| Grasping | 16.0 | 15.8 | 0.858 | –1.82–2.17 |
| Elbow flexors | 24.3 | 23.0 | 0.641 | –4.12–6.55 |
| Knee flexors | 15.0 | 17.1 | 0.126 | –4.81–0.64 |
| Knee extensors | 12.9 | 14.0 | 0.514 | –4.51–2.33 |
Fig. 1. Association of muscle strength and SMN2 copies in our study. Dashed line: patients with 2 SMN2 copies (n = 5). Complete line: patients with 3–4 SMN2 copies (n = 19).
DISCUSSION
Muscle strength was evaluated in 24 patients with chronic SMA who were followed up for a period of 12 months. Association with gender, age, SMA type, SMN2 copy number and ability to walk was established.
The muscle strength of patients with SMA yielded values that were lower than those for the normal population. In line with reports from other authors (10, 17) we found that knee extensors constituted the most deficient muscle group. This contrasts strongly with the normal population, where knee extensors are the most powerful muscle group (18).
As expected, the most significant differences we found were between ambulatory and non-ambulatory patients. Muscles of lower and upper extremities were stronger in the ambulatory patients than in the non-ambulatory patients. Knee extensors were 4 times stronger in patients who could walk. Nevertheless, as regards normal values (15, 19) for the age of ambulatory patients, their strength was markedly reduced. In this group, the strength values for grasping and elbow flexors were 3–7 times higher than the values for knee flexors and extensors. This confirms the greater impact of the disease in proximal muscles and the lower extremities, even in children who maintain the ability to walk (5).
In evaluating patients with SMA type III who had lost their ability to walk prior to the study, we found that their muscle strength was lower than that of those who were able to walk. The differences were statistically significant (Table III). Disease severity therefore appears to have a greater relationship with the ability to walk than with the SMA type, as noted by Kroksmark et al. (19). Indeed, in our study, patients who had formerly been ambulatory showed very low values of muscle strength once they had lost their ability to walk, similar to those of patients who had never walked.
In healthy populations it has been reported that muscle strength values are 60% lower in females than in males, except for the 12–14-year-old group, where girls show greater strength than do boys (15, 19). We found no significant differences between the sexes, nor did we find differences related to age; that is, between younger patients (children and teenagers) and adults. In our study there were only 3 patients who were able to walk, so no comparisons could be made in this regard. Nevertheless, our findings are consistent with those of Merlini et al. (10) in a study of 33 patients with SMA, of whom only 5 could walk.
A stable clinical picture has been described, mainly among patients with type II SMA (20). No statistically significant changes were observed in the results for muscle strength between the initial testing and the end of our study. Nevertheless, one year is insufficient to draw definite conclusions about the stability of the disease.
As for the measurement of manual grasping in young children, difficulty was noted in adapting the hand to the dynamometer. This could impede correct measurement. Molenaar et al. (21) have discussed this issue, validating another type of myometer. Major joint deformities of the forearm and the wrist also made positioning for correct measurement more difficult. These factors should be taken into account in future studies. However, we were able to make objective measurements and to confirm that patients with SMA do indeed undergo a considerable reduction in muscle strength. We found extremely low values, mainly in the knee and elbow extensors, as has been observed by other authors (10, 22, 23).
A recent report revealed a better functional level, measured with the Hammersmith scale, in type II patients with 3 SMN2 copies than in type II patients with 2 SMN2 copies (24). In line with these findings, our study detected higher strength values in the group of 3 SMN2 copies despite the fact that our series included type II and type III patients. These results provide further evidence to support the influence of SMN2 copies in SMA evolution and should be taken into account when selecting patients for clinical trials.
Our findings support the use of HD as a practical, reliable and affordable tool to quantify muscle weakness in patients with SMA, even in those with very serious deficits. In our experience HD allows a more objective measure than MRC, given that small changes in muscle strength can be documented. Measuring muscle strength by means of HD can be particularly helpful for monitoring the progression of SMA and for evaluating the results of therapeutic trials.
REFERENCES