


Sei-ichiro Motegi1, Yoko Yokoyama1, Sachiko Ogino1, Kazuya Yamada1, Akihiko Uchiyama1, Buddhini Perera1, Yuko Takeuchi1, Hiroshi Ohnishi2 and Osamu Ishikawa1
1Department of Dermatology, Gunma University Graduate School of Medicine, Maebashi, Japan, and 2Department of Laboratory Sciences, Gunma University Graduate School of Health Sciences, Maebashi, Japan
LEOPARD syndrome (LS) is an autosomal dominant condition with multiple anomalies, including multiple lentigines. LS is caused by mutations in PTPN11, encoding the protein tyrosine phosphatase, SHP-2. We report here 2 unrelated Japanese cases of LS with different PTPN11 mutations (p.Y279C and p.T468P). To elucidate the pathogenesis of multiple lentigines in LS, ultrastructural and immunohistochemical analyses of lentigines and non-lesional skin were performed. Numerous mature giant melanosomes in melanocytes and keratinocytes were observed in lentigines. In addition, the levels of expression of endothelin-1 (ET-1), phosphorylated Akt, mTOR and STAT3 in the epidermis in lentigines were significantly elevated compared with non-lesional skin. In in vitro assays, melanin synthesis in human melanoma cells expressing SHP-2 with LS-associated mutations was higher than in cells expressing normal SHP-2, suggesting that LS-associated SHP-2 mutations might enhance melanin synthesis in melanocytes, and that the activation of Akt/mTOR signalling may contribute to this process. Key words: LEOPARD syndrome; SHP-2; multiple lentigines; melanocyte; mTOR signalling.
Accepted Apr 27, 2015; Epub ahead of print xx
Acta Derm Venereol 2015; XX: XX–XX.
Sei-ichiro Motegi, Department of Dermatology, Gunma University Graduate School of Medicine, 3-39-22 Showa-machi, Maebashi, Gunma, 371-8511, Japan. E-mail: smotegi@gunma-u.ac.jp
LEOPARD syndrome (LS) (OMIM# 151100) is an autosomal dominant condition. The acronym LEOPARD indicates that the condition is associated with multiple Lentigines, Electrocardiographic conduction defects, Ocular hypertelorism, Pulmonary stenosis, Abnormalities of the genitalia, Retardation of growth, and sensorineural Deafness (1). Patients with LS carry heterozygous mutations in the protein-tyrosine phosphatase non-receptor type 11 (PTPN11) gene located on chromosome 12q24.1, which encodes the protein tyrosine phosphatase, SHP-2 [Src homology 2 (SH2)-containing protein-tyrosine-phosphatase (PTP)] (2). SHP-2 plays an important role in mediating multiple downstream biological responses, including proliferation, adhesion and migration (3–5). SHP-2 is a ubiquitously expressed, cytoplasmic PTP bearing 2 SH2 domains at its N-terminal end (Fig. 1q). In the basal state, the N-terminal SH2 domain interacts with the PTP domain, resulting in auto-inhibition of PTP activity. The binding of SHP-2 via its SH2 domains to tyrosine-phosphorylated growth factor receptors or docking proteins results in disruption of this intramolecular interaction, leading to exposure of the PTP domain and catalytic activation (3). These activated SHP-2 induce the dephosphorylation of adaptor proteins, including GRB2-associated binder 1 (GAB1), resulting in the regulation of downstream signalling, such as RAS/MAPK and phosphoinositide 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) signalling cascades (3, 6, 7). Disorders with RAS/MAPK activation caused by mutations have recently been called “RASopathies”, and include Noonan syndrome, neurofibromatosis 1 and LS (6–9). It has been reported that LS SHP-2 mutants bind preferentially to upstream activators to prolong substrate turnover, thus engendering gain-of-function phenotypes (7). It has also been reported that PTPN11 mutation Y279C knockin mice, which have both endogenous WT SHP-2 and SHP-2 with Y279C mutation, recapitulated the human disorder, with short stature, craniofacial dysmorphia and morphological, histological, echocardiographic and molecular evidence of hypertrophic cardiomyopathy (10). However, the mechanism by which SHP-2 mutation causes multiple lentigines is not well understood. To assess whether there are abnormalities of melanogenesis in melanocytes and/or melanosome transfer to keratinocytes, we electron-microscopically examined the melanin/melanosome organization in melanocytes and keratinocytes in lentigines from 2 patients treated in our institution. In addition, immunohistochemical staining for endothelin-1 (ET-1), α-melanocyte stimulating hormone (α-MSH), stem cell factor (SCF), phosphorylated STAT-3, phosphorylated Akt and phosphorylated mTOR in the lentigines and non-lesional skin from our patients was analysed. The effects of LS-associated SHP-2 mutations on melanin synthesis in human melanoma cells in vitro were also analysed.
MATERIALS AND METHODS
Clinical examination
Skin specimens were obtained from the patients, and age- and sex-matched healthy volunteers. This study was approved by the local research ethics committee of Gunma University. Patients provided written informed consent before participation. This study was conducted in accordance with the principles of the Declaration of Helsinki.
Genetic analysis
Genomic DNA was isolated from whole blood cells using Wizard® Genomic DNA Purification Kit (Promega, Madison, WI, USA). Genomic DNA was amplified for the 15 exons and flanking introns of the PTPN11 gene by polymerase chain reaction (PCR) with 15 sets of primers, as described previously (11). The PCR products were directly sequenced using an ABI Prism 3100 sequence analyser (Applied Biosystems, Foster City, CA, USA).
Ultrastructural and immunohistochemical analysis
For electron microscopy, skin tissues were fixed 1% glutaraldehyde, subsequently fixed with osmium tetraoxide (OsO4), then dehydrated in an ethanol series and embedded in resin.
Human tissue was embedded in paraffin, and sectioned 3 μm thick. The sections were deparaffinized with xylene for 30 min. Tissue sections were treated for antigen retrieval with a pressure cooker for 10 min at 120°C. After blocking using Peroxidase Blocking (Dako, Copenhagen, Denmark) for 5 min, and Protein Block (Dako) for 10 min, sections were incubated with anti-Melan-A antibody (Abcam, Cambridge, UK), anti-endothelin-1 antibody (Abcam), anti-αMSH (Acris Antibodies, San Diego, CA, USA), anti-SCF antibody (Abcam), anti-phospho-Akt (Ser473) antibody, anti-phospho-mTOR (Ser2448) antibody (Cell Signaling, Danvers, MA, USA), or anti-phospho-STAT-3 (Ser727) antibody (Millipore, Billerica, MA, USA) overnight at 4°C. After washing, the sections were incubated with a horseradish peroxidase-labelled polymer-conjugated anti-mouse secondary antibody (ENVISION+; Dako) for 1 h at room temperature. Finally, colour was developed with 3,3′-diaminobenzidine tetrahydrochloride.
Transfection and melanin analysis
To construct expression vectors for LS-associated mutants (Y279C, T468P) of SHP-2, partial cDNA fragments were amplified by PCR using primers containing nucleotide substitutions, corresponding to Y279C or T468P mutation. A cDNA clone for human SHP-2 (Clone #FLJ75588AAAN, National Institute of Technology and Evaluation, Biological Resource Center) was used as a template for the reaction. PCR products were then assembled and cloned into pCAGGS plasmid (kindly provided by J. Miyazaki (Osaka University)) by the use of In-Fusion cloning system (Clontech , Mountain View, CA). In all constructs, enhanced green fluorescent protein (EGFP) was fused to the C-terminus of the mutant SHP-2. An expression vector for WT SHP-2 was also constructed using the same procedure. DNA sequences of all constructs were confirmed by DNA sequence analyses. These plasmids were transfected into human melanoma cells G-361 (JCRB Cell Bank, Osaka, Japan) using X-tremeGENE HP DNA Transfection Regent (Roche Diagnostics, Indianapolis, IN, USA) according to the manufacture’s protocol. Forty-eight hours after transfection, incubated cells were homogenized and the amount of melanin in melanoma cells was determined by a Human Melanin enzyme-linked immunosorbent assay (ELISA) Kit (Cusabio, Hubei Province, China) according to the manufacture’s protocol. To analyse the effect of mTOR inhibitor, rapamycin (Sigma, St. Louis, MO) on the melanin synthesis, transfected cells were treated with 50 nM rapamycin for 48 h and then melanin synthesis was analysed using ELISA kit.
Western blotting
Transfected human melanoma cells G-361 were incubated in 10% foetal bovine serum (FBS)-containing medium for 48 h. After washing with ice-cold phosphate-buffered saline (PBS), cells were disrupted in lysis buffer (20 mM Tris-HCL (pH 7.6), 140 mM NaCl, 1% Nonidet P-40) containing 1 mM phenylmethylsulfonyluoride and aprotinin (10 mg/ml) on ice. Lysates were centrifuged at 10,000 × g for 15 min at 4°C and the resulting supernatants were subjected to sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE), followed by immunoblot analysis using rabbit anti-SHP-2 antibody or mouse anti-actin antibody (Millipore). Horse-radish peroxidase (HRP)-conjugated anti-rabbit immunoglobulin G (IgG) antibodies (Jackson ImmunoResearch, West Grove, PA, USA) were used with enhanced chemiluminescence (ECL) (Thermo Scientific, Rockford, IL, USA) to image immunoblots.
Statistical analysis
p-values were calculated by 1-way analysis of variance (ANOVA) followed by Bonferroni’s post-hoc test. Error bars represent standard errors of the mean, and numbers of experiments (n) are as indicated.
RESULTS
Clinical and histological features of 2 cases of LEOPARD syndrome
Case 1. A 23-year-old Japanese female had multiple lentigines at birth, and the number of lentigines gradually increased as she aged. She developed sensorineural deafness at 2 years of age, and was noted to have hypertrophic cardiomyopathy at 12 years of age. Her parents were non-consanguineous, and her family history was unremarkable. Physical examination revealed ocular hypertelorism and multiple light- or dark-brown lentigines of varying sizes over her whole body (Fig. 1a–c). There was no pulmonary stenosis, abnormalities of the genitalia or retardation of growth.
Histopathological examination of lentigo on her back (Fig. 1c) showed a slight degree of acanthosis with an increased amount of melanin deposition in the epidermis (Fig. 1d). In contrast, histopathological examination of non-lesional skin on her back showed no basal pigmentation (Fig. 1g). To examine the number and localization of melanocytes, melanocyte marker, melan-A, also known as melanoma antigen recognized by T cells 1 (MART-1), was stained. The melan-A staining demonstrated an increased number of melanocytes in the epidermis in the lentigo (Fig. 1e) compared with that in non-lesional skin (Fig. 1h). Interestingly, melan-A staining in the sweat glands was observed in both lentigo and non-lesional skin (Fig. 1f, i), suggesting that melanocytes might have existed in the sweat glands in this patient.
Case 2. A 14-year-old Japanese female had multiple lentigines at birth, which gradually spread over her whole body. Her parents were non-consanguineous, and her family history was not contributory. Physical examination revealed ocular hypertelorism and multiple light- or dark-brown lentigines of varying sizes over her whole body (Fig. 1j–l). An electrocardiogram showed right axis deviation. There was no pulmonary stenosis, abnormalities of the genitalia, retardation of growth and sensorineural deafness. Histopathological examination of lentigo on her back (Fig. 1l) showed the elongation of rete ridges and pigment accumulation from the basal to upper layers of the epidermis (Fig. 1m). Melan-A staining showed an increased number of melanocytes in the epidermis (Fig. 1n). No melan-A staining was observed in the sweat glands in the dermis (Fig. 1o).
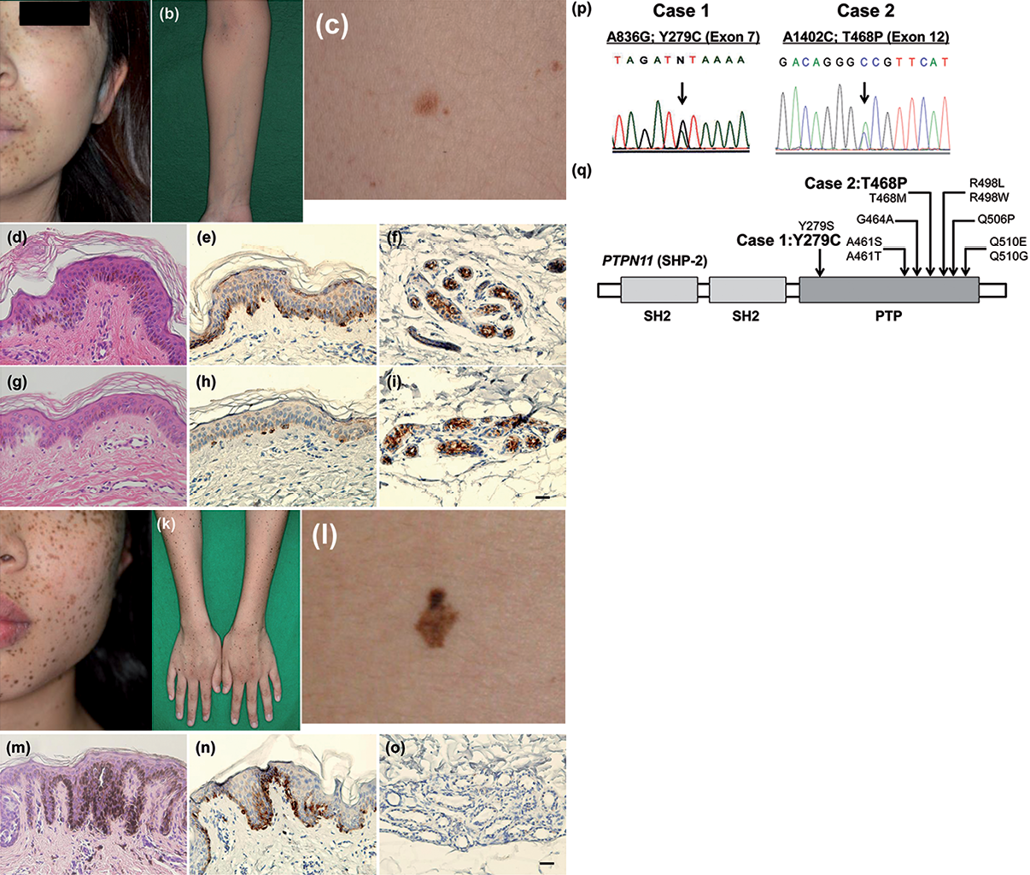
Fig. 1. Clinical, histopathological and genetic features of the LEOPARD syndrome patient. Case 1. (a–c) Multiple light- or dark-brown lentigines of varying sizes over her whole body. (d) Histopathological examination of lentigo on her back showing. Acanthosis with an increased amount of melanin deposition in the epidermis. (e) Melan-A staining showed an increased number of melanocytes in the epidermis and in (f) the sweat glands in the lentigo. (g) Results of the histopathological examination of non-lesional skin on the back. No basal pigmentation. (h, i) Melan-A staining of the epidermis and sweat glands in the non-lesional skin. Case 2. (j–l) Multiple light- or dark-brown lentigines of varying sizes over her whole body. (m) Histopathological examination of lentigo on her back showing (l) elongation of rete ridges and pigment accumulation from the basal to upper layers of the epidermis. (n) Melan-A staining showed an increased number of melanocytes in the epidermis in the lentigo but (o) no staining was present in the sweat glands. Scale bar = 20 μm (p). PTPN11 mutation analysis. Case 1 (left-hand panel): there was a heterozygous PTPN11 c.836A>G mutation in exon 7, predicted to cause a change of tyrosine 279 to cysteine at the protein level (p.Y279C). Case 2 (right-hand panel): there was a heterozygous PTPN11 c.1402A>C mutation in exon 12, which was predicted to cause a change of threonine 468 to proline at the protein level (p.T468P). (q) Distribution of mutations of the PTPN11 gene in LEOPARD syndrome.
PTPN11 mutation analysis
In case 1, we identified a heterozygous PTPN11 c.836A>G mutation in exon 7, predicted to cause the change of tyrosine 279 into cysteine at the protein level (p.Y279C) (Fig. 1p: left). In case 2, we identified a heterozygous PTPN11 c.1402A>C mutation in exon 12, predicted to cause a change of threonine 468 into proline at the protein level (p.T468P) (Fig. 1p: right). No mutations in the PTPN11 gene were found in parents of the patient in case 2. Based on the clinical and genetic findings, a diagnosis of LS with a PTPN11 mutation was established in both cases. The distribution of mutations of the PTPN11 gene in LEOPARD syndrome is shown in Fig. 1q.
Ultrastructural analysis of lentigines
There have been 2 reports of ultrastructural analyses in lentigines in LS patients (12, 13). Bhawan et al. (12) reported that giant melanosomes were found in the lentigines in LS, and immature melanosomes are present in the keratinocytes of LS. In addition, Fryer et al. reported the electronmicroscopic findings of large accumulations of melanin within Langerhans cells in the lentigines of LS (13). However, the precise characteristics of melanin/melanosomes in melanocytes and keratinocytes in the LS have not been fully elucidated. Therefore, we performed ultrastructural analyses of the lentigo and non-lesional skin in our 2 patients by electron microscopy.
In the lesional skin, many mature (stage III–IV) melanosomes were observed in melanocytes, and the diameter of the melanosomes in melanocytes was 250–450 nm (Fig. 2; left and middle panels of MC/KC and MC). In contrast, normal numbers and sizes (200–230 nm) of melanosomes in melanocytes were observed in the non-lesional skin of case 2 (Fig. 2: right panels of MC/KC and MC). The number of melanosomes in melanocyte in lesional skin was higher than that in non-lesional skin. Regarding the melanosomes in keratinocytes, numerous melanosomes were observed in the keratinocytes around the melanocytes in lesional skin, and most of them were fused with each other, and 5–8 melanosomes formed compound melanosomes (Fig. 2; left and middle panels of MC/KC). The diameter of these compound melanosomes in keratinocytes was 450–500 nm. In contrast, in the non-lesional skin of case 2, normal-sized compound melanosomes (approximately 350 nm) were observed in the keratinocytes (Fig. 2; right panel of MC/KC). We could not find melanosomes in the Langerhans cells in the lentigo in LS case (Fig. 2; LC). These results suggest that an abnormal increase in the production of melanin/melanosomes in melanocytes might be associated with the pathogenesis of LS.
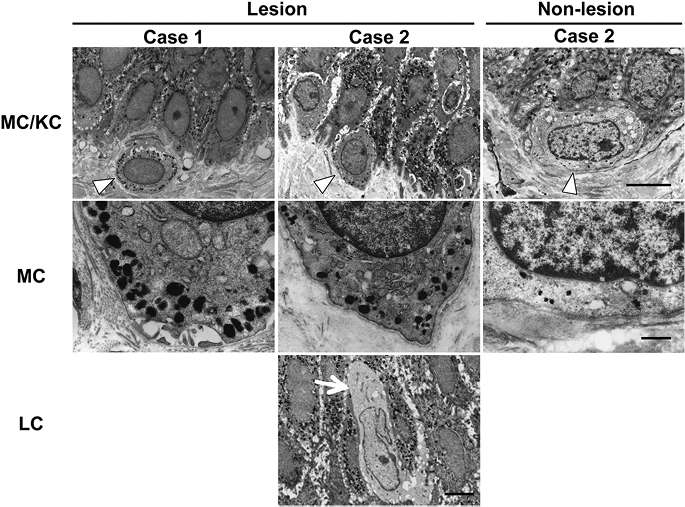
Fig. 2. Ultrastructural analysis of lentigines. In lesional skin, many mature melanosomes in melanocytes (MC) (arrowhead) and keratinocytes (KC). The diameter of the melanosomes in MC was 250–450 nm. The diameter of compound melanosomes in KC was 450–500 nm (left-hand and middle panels of MC/KC and MC). The normal number and size (200–230 nm) of melanosomes in MC (arrowhead) in non-lesional skin of LS (right-hand panels of MC/KC and MC). No melanosomes were noted in Langerhans cells (LC) (arrow) in the lentigo in LEOPARD syndrome. (Upper panels: scale bar = 5 μm. Middle panels: scale bar = 1 μm. Lower panel: scale bar = 5 μm.)
Immunohistochemical analysis of lentigines
Several cytokines and growth factors, such as ET-1, αMSH and SCF, which are secreted from keratinocytes, can induce the migration, proliferation and melanogenesis of melanocytes, and the increased secretion of these factors is involved in the hyperpigmentation in lentigo seniles and other pigmentation-associated disorders (14, 15). To assess the roles of ET-1, αMSH and SCF in the pathogenesis of lentigines in LS, immunohistochemical staining of lesional and non-lesional skin was performed. In the immunohistochemical analyses, the staining of ET-1 in the epidermis in lentigines was significantly enhanced compared with that in non-lesional skin and healthy control skin (Fig. 3a). However, the staining of α-MSH and SCF revealed weakly positive staining throughout the epidermis in both the lentigo and non-lesional skin, and there was no distinct difference (Fig. 3a).
LS mutations in SHP-2 are located in the PTP domain, involving the catalytic domain, resulting in decreased phosphatase activity, and subsequently induced the activation of Akt/mTOR signalling, STAT3/JNK1/2 signalling and FAK signalling in in vitro assays (6, 7, 10). Therefore, we examined the activation of Akt, mTOR and STAT3 in the epidermis of the lentigo and non-lesional skin in our LS patients. The nuclear staining of phosphorylated Akt, mTOR and STAT3 was observed throughout the epidermis in the lentigo in our LS patients (Fig. 3b). The staining for activated Akt, mTOR and STAT3 in the lesional skin was observed more than that in non-lesional skin and in skin from normal individuals. These results suggest that the activation of the Akt/mTOR and STAT3 pathways might be involved in the pathogenesis of multiple lentigines in LS.
Effect of LS-associated SHP-2 mutations on melanin synthesis in human melanoma cells in vitro
Finally, we examined melanin synthesis in human melanoma cells overexpressing SHP-2 with LS-associated mutations in vitro. In immunoblotting, EGFP-fused wild-type (WT) SHP-2 and SHP-2 with LS-associated mutations (T468P or Y279C) were detected (Fig. 3c: lanes 2–4). Endogenous SHP-2 was detected in all melanoma cells (Fig. 3c: lanes 1–4). These results confirmed that WT SHP-2 and SHP-2 with LS-associated mutations (T468P or Y279C) were successfully transfected into human melanoma cells 48 h after transient transfection. The amounts of melanin synthesis in the melanoma cells overexpressing WT SHP-2 or SHP-2 with LS-associated mutations (T468P or Y279C) were significantly higher than that in control melanoma cells (Fig. 3d). In addition, the amount of melanin in human melanoma cells overexpressing LS-associated SHP-2 mutation (T468P) was significantly higher than that in cells overexpressing WT SHP-2. There was a tendency that the amount of melanin in melanoma cells overexpressing LS-associated SHP-2 mutation (Y279C) was higher than that in cells overexpressing WT SHP-2. We also found that mTOR inhibitor, rapamycin, significantly reversed the enhancements of melanin synthesis in SHP-2 with LS-associated mutations (T468P or Y279C) transfected human melanoma cells (Fig. 3d), suggesting that the activation of mTOR signalling in melanocytes might be involved in the enhancement of melanin synthesis by overexpression of SHP-2 with LS-associated mutations.
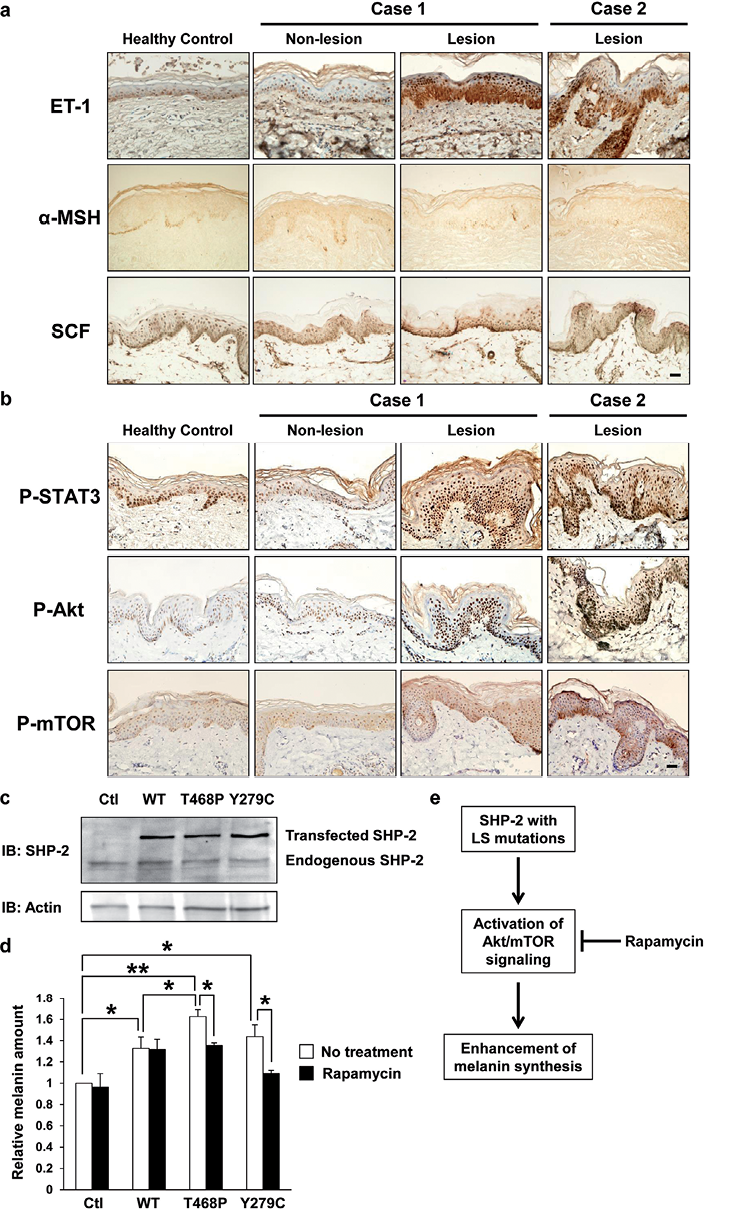
Fig. 3. (a) Immunohistochemical staining for endothelin-1 (ET-1), α-melanocyte stimulating hormone (α-MSH) and stem cell factor (SCF) in the epidermis in lentigines and non-lesional skin. Staining of ET-1 in epidermis in the lentigines was significantly enhanced. Staining of α-MSH and SCF was weakly positive throughout the epidermis in both the lentigo and non-lesional skin, and there was no difference between lesion and non-lesion. Scale bar = 20 μm. (b) Immunohistochemical staining for phosphorylated STAT3 (P-STAT3), phosphorylated Akt (P-Akt) and phosphorylated mTOR (P-mTOR) in the epidermis in lentigines and non-lesional skin. Staining of P-STAT3, P-Akt and P-mTOR in epidermis in the lentigines was significantly enhanced. Scale bar = 20 μm. (c, d) The effects of LEOPARD syndrome (LS)-associated SHP-2 mutations on the melanin synthesis in human melanoma cells in vitro. (c) Protein levels of wild-type (WT) SHP-2 and SHP-2 with LS-associated mutations (T468P or Y279C) in human melanoma cells detected by immunoblotting. At 48 h after transfection, the transfected cells were subjected to sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE), followed by immunoblotting with anti-SHP-2 and actin antibodies. (d) The relative quantification of the amount of melanin in melanoma cells transfected with WT SHP-2 or SHP-2 with LS-associated mutations (T468P or Y279C) with or without rapamycin (50 nM) at 48 h after transfection. All values represent means ± SEM. **p < 0.01, *p < 0.05, n = 3. (e) Schematic representation for the role of SHP-2 with LS-associated mutations in melanin synthesis in melanocytes. SHP-2 with LS-associated mutations enhances melanin synthesis in melanocytes, and the activation of Akt/mTOR signalling contributes to this process.
DISCUSSION
Genetic analysis of our patients revealed a heterozygous point mutation in exon 12, p.T468P, and in exon 7, p.Y279C, respectively. To the best of our knowledge, there has been only one reported Japanese case with the same mutation (p.T468P) found in case 2 (16). In this Japanese patient, malignant melanoma with a somatic BRAF mutation developed at the age of 60 years. In addition to this case, there was another case of LS associated with malignant melanoma with a PTPN11 mutation (pY279C) (17). Since both of our cases had a mutation of PTPN11 (p.T468P or p.Y279C) associated with the possible development of malignant melanoma, periodic observation of the whole body will be needed.
We could not analyse the loss of heterozygosity (LOH) in lentigines due to the small size of the sample limitation. In addition, we found no published evidence that LOH is associated with the pathogenesis for causing lentigines. Analysis of LOH in granular cell tumours in LS patient has been reported, and the absence of LOH of PTPN11 supports published functional data that T468M is a dominant-negative mutation (18). In addition, LOH was not detected in malignant melanoma in an LS patient with T468P SHP-2 mutant (16). Based on these findings, we speculate that LOH might not be associated with the pathogenesis of lentigines.
Stewart et al. (19) demonstrated that a SHP-2 mutation of LS induced the migration of neural crest cells and prevented the apoptosis of neural crest cells, resulting in hyperpigmentation of zebrafish in which mRNA encoding LS mutation was injected. Since the melan-A staining demonstrated an increased number of melanocytes in the epidermis in the lentigo in our cases, LS-associated mutants (Y279C, T468P) of SHP-2 may account for the enhancement of migration and the prevention of the apoptosis of melanocytes. In our patient (case 1), melan-A staining was observed in sweat glands, suggesting that melanocytes may migrate to the wrong place during development. However, melan-A staining was not observed in the sweat glands in patient 2; therefore, further examination is needed to confirm this finding.
Our findings in the ultrastructural analyses of lentigo showed enhanced melanin/melanosomes in melanocytes, suggesting that abnormal synthesis of melanosomes in melanocytes caused by a SHP-2 mutation might also be involved in the pathogenesis of multiple lentigines of LS. This is the first detailed ultrastructural analysis to examine the characteristics of melanin/melanosomes in melanocytes and keratinocytes in lentigines and normal skin in patients with LS. Bhawan et al. (12) reported that giant melanosomes were observed in lentigines in LS, and immature melanosomes were present in the keratinocytes of LS. In our patient, numerous giant melanosomes were observed in melanocytes; however, mature melanosomes were observed in both melanocytes and keratinocytes. Fryer et al. reported electron-microscopic findings of large accumulations of melanin within Langerhans cells in the lentigines of LS (13). However, this finding was not identified in our analysis.
In the immunohistochemical analyses, the staining of ET-1 in the epidermis in lentigines was significantly enhanced compared with that in non-lesional skin and healthy control skin. Kadono et al. (20) previously reported that ET-1 expression was increased in epidermis in lentigo in healthy individuals, suggesting that the enhancement of ET-1 expression in epidermis in lentigo might not be a specific change in the lentigines of LS. In addition, ET-1 expression was not enhanced in keratinocytes in non-lesional area with the mutation of PTPN11, suggesting that PTPN11 mutation in keratinocytes does not directly cause the enhancement of the secretion of ET-1 from keratinocytes. These findings suggest that melanocytes and/or keratinocytes with PTPN11 mutation of LS may secrete some cytokines and/or growth factors, and this cell autonomous mechanism may result in the enhancement of ET-1 secretion from keratinocytes and the development of lentigines in LS. Further research is needed to identify the role of ET-1 in the pathogenesis of lentigines in LS.
The staining of phosphorylated Akt, mTOR and STAT3 in the epidermis, including keratinocytes and melanocytes, in lentigo lesions was enhanced compared with the levels in non-lesional skin and skin from normal individuals. Consistent with our present results, the activation of the Akt/mTOR and STAT3 pathways in LS-mutant (Y279C or T468M)-transfected cells was enhanced by stimulation with epidermal growth factor (EGF) or insulin growth factor-1 (IGF-1) in vitro (10, 21). In addition, the activation of mTOR is strongly associated with the development of malignant melanoma in vivo (22, 23), and treatment with a mTOR inhibitor, rapamycin, reversed the cardiac defects in LS-mutant SHP-2 (Y279C) knockin mice (10), suggesting that the activation of mTOR in keratinocytes and melanocytes may be involved in the pathogenesis of the multiple lentigines in LS.
With respect to SHP-2 and the functions of melanocytes, it has been reported that blockade of SHP-2 activity abrogated MET-dependent activation of Erk1/2 and Akt in melanocytes (24), suggesting that SHP-2 might regulate the functions of melanocytes, such as proliferation and migration. However, the role of SHP-2 with LS-associated mutations in melanin synthesis had not yet been characterized. This is the first study to investigate the role of LS-associated SHP-2 mutations in melanin synthesis in vitro. We show a schematic representation for the role of SHP-2 with LS-associated mutations in melanin synthesis in melanocytes in Fig. 3e. We demonstrated that melanin synthesis was significantly increased in human melanoma cells overexpressing SHP-2 with LS-associated mutations. It has been reported that activation of mTOR is associated with melanogensis and the pathogenesis of malignant melanoma (22, 23). We showed that mTOR inhibitor, rapamycin, reversed the enhancements of melanin synthesis in SHP-2 with LS-associated mutations (T468P or Y279C) transfected human melanoma cells. These results suggest that mTOR signalling might be involved in the mechanism(s) underlying LS-associated SHP-2 mutant-induced hypermelanogenesis, and that rapamycin might have therapeutic potential for multiple lentigines in patients with LS.
The authors declare no conflicts of interest.
REFERENCES

